Проте лише близько 2% медичних показань, що відповідають специфічним потребам хворих з орфанними захворюваннями, охоплені схваленими лікарськими засобами. Про це заявляє, наприклад, німецька Асоціація науково-дослідних фармацевтичних компаній (Die forschenden Pharma-Unternehmen — vfa) у своєму пресрелізі від 26 лютого 2024 р.
В якості прикладу можна привести нещодавнє, у лютому 2024 р., схвалення препарату для лікування бічного аміотрофічного склерозу (БАС) — рідкісного захворювання, що призводить до інвалідизації та смерті. До цього схвалення в ЄС для лікування БАС був дозволений лише один лікарський засіб — рилузол. Проте новий препарат — тоферсен, спрямований на генетичну мутацію (із виробленням дефектних ферментів SOD1), з якою живуть лише 2% хворих із цим захворюванням. Тобто для 98% інших пацієнтів із схваленням тоферсену принципово нічого не змінилося.
Про значну невдоволену потребу свідчить те, що майже третина орфанних препаратів, дозволених з 2010 р., стала першим варіантом лікування за схваленим показанням; у всіх інших випадках Європейське агентство з лікарських засобів (European Medicines Agency — EMA) підтвердило, що орфанні ліки мали клінічно значущу перевагу або зробили значний внесок у догляд за пацієнтами, тобто забезпечили «значну користь» (рис. 1).
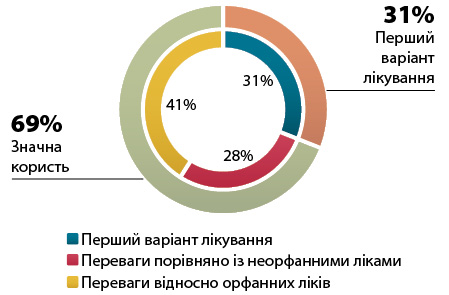
Майже 25 років Регламенту з орфанних препаратів
Проте ще чверть століття тому ситуація із доступністю ліків була набагато гіршою. Як нагадує Європейська федерація асоціацій фармацевтичної промисловості (Еuropean Federation of Pharmaceutical Industry Associations — EFPIA), до 2000 р. для 6 тис. виявлених рідкісних захворювань загалом було доступно лише 8 препаратів.
Європейський регламент щодо орфанних лікарських засобів (Regulation (EC) № 141/2000) став поворотним пунктом в історії розробки засобів для лікування рідкісних захворювань. Ним передбачено кілька заходів, зокрема підвищення експертного потенціалу, зменшення регулятивних зборів, стимулів для малих і середніх підприємств і 10-річний термін ексклюзивності на ринку, який за певних обставин можна подовжити на 2 роки.
Цей підхід працює. У період з 2000 до 2018 р. Регламент сприяв наданню статусу «орфанні ліки» понад 2700 разів, завдяки чому, у свою чергу, видано понад 230 дозволів на маркетинг для лікування близько 90 рідкісних захворювань.
Подальший прогрес під питанням
На жаль, існує реальний ризик повернути назад на шляху до сталого розвитку, попереджає EFPIA. У квітні 2023 р. у контексті перегляду фармацевтичного законодавства Європейська комісія запропонувала переглянути систему стимулів для ліків-сиріт. Зокрема, скоротити тривалість ринкової ексклюзивності та запровадити більш суворі критерії для визнання орфанного статусу. Щоб інформаційно підкріпити обговорення цієї складної теми, EFPIA замовила звіт із моделюванням розвитку ситуації. За висновками аналітиків, запропоновані зміни негативно вплинуть на розвиток 45 продуктів у Європі в період між 2020 і 2035 р. Це означатиме зменшення кількості успішних інновацій на 12%, що потенційно позбавить близько 1,5 млн європейських пацієнтів із рідкісними захворюваннями нового варіанта лікування. Це також призведе до зменшення витрат на дослідження та розробки в Європі на 4,5 млрд євро.
Доступ до інновацій у межах ЄС відрізняється
Звичайно, вплив нових ліків залежить від того, чи отримали пацієнти доступ до них. У даних щорічного дослідження EFPIA «Patients W.A.I.T Indicator» (опублікований у квітні 2023 р.) виявлено, що у 2022 р. середній термін до відшкодування інноваційних ліків у країнах ЄС і Європейської економічної зони (ЄЕЗ) досяг 517 днів, коливаючись від 128 днів у Німеччині до 1351 дня на Мальті. За результатами аналізу основних причин EFPIA встановлено 10 факторів, які спричиняють затримки (таблиця).
| Група причин | Потенційні механізми |
| Ухвалення рішення про дозвіл на маркетинг | 1. Швидкість регуляторного процесу.
2. Доступність ліків до ухвалення рішень про їх схвалення. |
| Узгодження цін та відшкодування | 3. Ініціація процесу.
4. Швидкість національних процедур та їх дотримання. |
| Оцінка медичних технологій | 5. Вимоги до терапевтичної користі.
6. Вимоги до економічної ефективності. 7. Оцінка потреб цільової групи. |
| Ресурсна та фінансова забезпеченість системи | 8. Обсяг бюджету для запровадження інновацій.
9. Стан інфраструктури медичної допомоги та її доступність. |
| Ухвалення рішень на субнаціональному рівні | 10. Багатофакторний процес створення локальних формулярів та протоколів. |
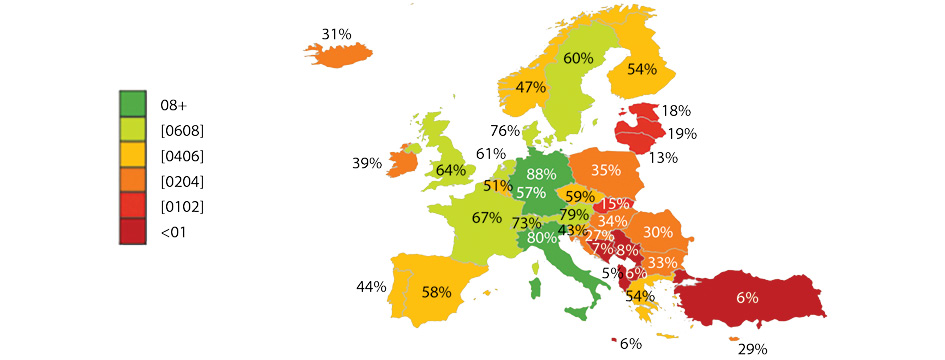
Зафіксовано деякі загальні закономірності: як правило, пацієнти в Північній і Західній Європі отримують доступ до нових методів лікування протягом 100–200 днів після отримання дозволу на маркетинг, тоді як пацієнти в Південній і Східній Європі чекають 600–1000 днів. Це означає, що в будь-який момент часу доступність ліків в Європі різко відрізняється. Наглядно це ілюструється індикатором «W.A.I.T.» (Waiting to Access Innovative Therapies), який використовує EFPIA в щорічних звітах (рис. 2).
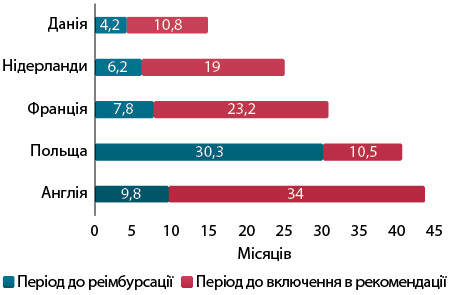
Зокрема, національні клінічні рекомендації (настанови) не завжди включають найновіші лікарські засоби. Унаслідок цього процес їх запровадження в клінічну практику розтягується в часі. Це досліджено на прикладі таргетних препаратів в онкології (рис. 3).
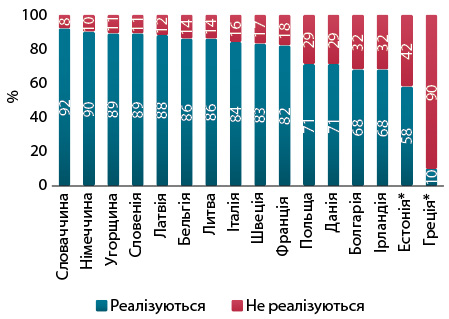
В інших випадках препарати є в переліках тих, що відшкодовують, але фінансування для їх застосування не спрямовується. Наприклад, пацієнти мають доступ лише до 76% продуктів, зареєстрованих у Румунії (рис. 4). Враховуючи цей досвід, не дивно, що не всі компанії вирішують подати заявку на відшкодування на подібних ринках.
Створення та підтримання екосистеми світового класу, у якій розробляються інноваційні ліки, прагне забезпечити європейський альянс EURORDIS-Rare Diseases Europe, який у ці дні запустив кампанію «Відстоювання рідкісних» (#ActRare2024). В її основі — дослідження «Rare 2030 foresight», виконане на замовлення Європейського парламенту на кошти, що виділені Єврокомісією. У ньому міститься заклик до лідерів ЄС вжити конкретних політичних заходів, щоб гарантувати, що 30 млн європейців із рідкісними захворюваннями матимуть довше та здоровіше життя.
www.eurordis.org, www.efpia.eu
Коментарі
Коментарі до цього матеріалу відсутні. Прокоментуйте першим